Yane's Site
My classwork from BIMM143
Class 17: Analyzing Sequencing Data
Yane Lee PID A17670350
Downstream Analysis
With each sample having its own directory containing the Kallisto output, we can import the transcript count estimates into R using:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("rhdf5")
Bioconductor version 3.22 (BiocManager 1.30.27), R 4.5.2 (2025-10-31)
Warning: package(s) not installed when version(s) same as or greater than current; use
`force = TRUE` to re-install: 'rhdf5'
library(tximport)
folders <- dir(pattern="SRR21568*")
samples <- sub("_quant","",folders)
files <- file.path(folders, "abundance.h5")
names(files) <- samples
txi.kallisto <- tximport(files, type="kallisto", txOut=TRUE)
1 2 3 4
head(txi.kallisto$counts)
SRR2156848 SRR2156849 SRR2156850 SRR2156851
ENST00000539570 0 0 0.00000 0
ENST00000576455 0 0 2.62037 0
ENST00000510508 0 0 0.00000 0
ENST00000474471 0 1 1.00000 0
ENST00000381700 0 0 0.00000 0
ENST00000445946 0 0 0.00000 0
We now have our estimated transcript counts for each sample in R. We can see how many transcripts we have for each sample:
colSums(txi.kallisto$counts)
SRR2156848 SRR2156849 SRR2156850 SRR2156851
2563611 2600800 2372309 2111474
And how many transcripts are detected in at least one sample:
sum(rowSums(txi.kallisto$counts)>0)
[1] 94561
We want to filter out the annotated transcripts with no reads
to.keep <- rowSums(txi.kallisto$counts) > 0
kset.nonzero <- txi.kallisto$counts[to.keep,]
keep2 <- apply(kset.nonzero,1,sd)>0
x <- kset.nonzero[keep2,]
Principal Component Analysis
We can now apply any exploratory analysis technique to this counts matrix. As an example, we will perform a PCA of the transcriptomic profiles of these samples.
pca <- prcomp(t(x), scale=TRUE)
summary(pca)
Importance of components:
PC1 PC2 PC3 PC4
Standard deviation 183.6379 177.3605 171.3020 1e+00
Proportion of Variance 0.3568 0.3328 0.3104 1e-05
Cumulative Proportion 0.3568 0.6895 1.0000 1e+00
Now we can use the first two principal components as a co-ordinate system for visualizing the summarized transcriptomic profiles of each sample:
plot(pca$x[,1], pca$x[,2],
col=c("blue","blue","red","red"),
xlab="PC1", ylab="PC2", pch=16)
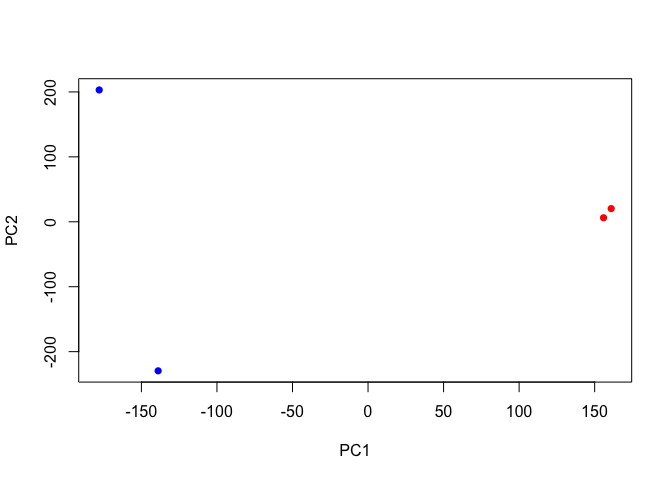
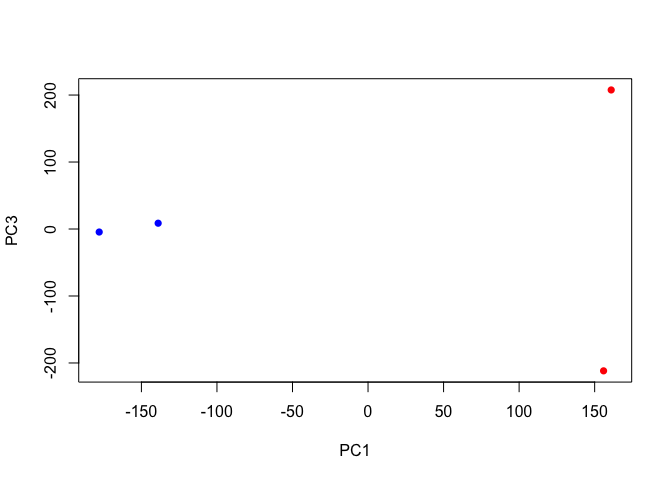
Q. Q. Use ggplot to make a similar figure of PC1 vs PC2 and a separate figure PC1 vs PC3 and PC2 vs PC3.
plot(pca$x[,1], pca$x[,3],
col=c("blue","blue","red","red"),
xlab="PC1", ylab="PC3", pch=16)
plot(pca$x[,2], pca$x[,3],
col=c("blue","blue","red","red"),
xlab="PC2", ylab="PC3", pch=16)
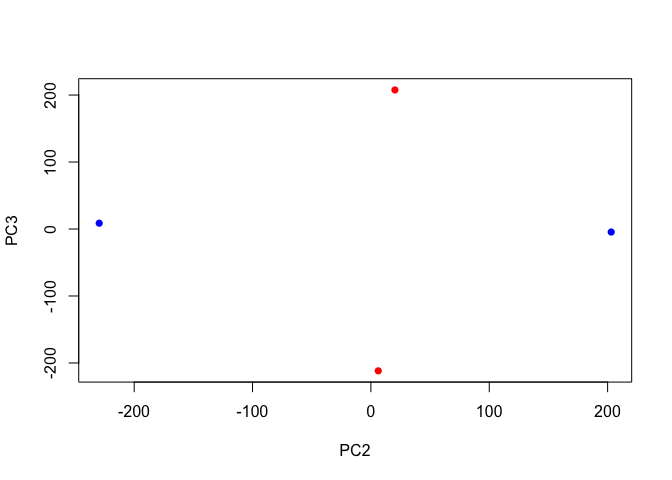
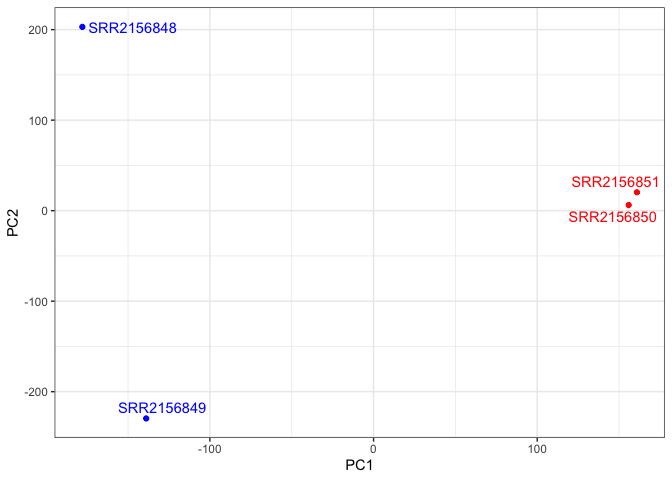
Plot with labels for the two control samples (SRR2156848 and SRR2156849) and the two enhancer-targeting CRISPR-Cas9 samples (SRR2156850 and SRR2156851).
library(ggplot2)
library(ggrepel)
mycols <- c("blue","blue","red","red")
ggplot(pca$x) +
aes(PC1, PC2, label=rownames(pca$x)) +
geom_point( col=mycols ) +
geom_text_repel( col=mycols ) +
theme_bw()
Differential-expression analysis
We can use DESeq2 to complete the differential-expression analysis that we are already familiar with:
library(DESeq2)
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: generics
Attaching package: 'generics'
The following objects are masked from 'package:base':
as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
setequal, union
Attaching package: 'BiocGenerics'
The following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabs
The following objects are masked from 'package:base':
anyDuplicated, aperm, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
unsplit, which.max, which.min
Attaching package: 'S4Vectors'
The following object is masked from 'package:utils':
findMatches
The following objects are masked from 'package:base':
expand.grid, I, unname
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: Seqinfo
Loading required package: SummarizedExperiment
Loading required package: MatrixGenerics
Loading required package: matrixStats
Attaching package: 'MatrixGenerics'
The following objects are masked from 'package:matrixStats':
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVars
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'Biobase'
The following object is masked from 'package:MatrixGenerics':
rowMedians
The following objects are masked from 'package:matrixStats':
anyMissing, rowMedians
sampleTable <- data.frame(condition = factor(rep(c("control", "treatment"), each = 2)))
rownames(sampleTable) <- colnames(txi.kallisto$counts)
dds <- DESeqDataSetFromTximport(txi.kallisto,
sampleTable,
~condition)
using counts and average transcript lengths from tximport
dds <- DESeq(dds)
estimating size factors
using 'avgTxLength' from assays(dds), correcting for library size
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
-- note: fitType='parametric', but the dispersion trend was not well captured by the
function: y = a/x + b, and a local regression fit was automatically substituted.
specify fitType='local' or 'mean' to avoid this message next time.
final dispersion estimates
fitting model and testing
res <- results(dds)
head(res)
log2 fold change (MLE): condition treatment vs control
Wald test p-value: condition treatment vs control
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENST00000539570 0.000000 NA NA NA NA
ENST00000576455 0.761453 3.155061 4.86052 0.6491203 0.516261
ENST00000510508 0.000000 NA NA NA NA
ENST00000474471 0.484938 0.181923 4.24871 0.0428185 0.965846
ENST00000381700 0.000000 NA NA NA NA
ENST00000445946 0.000000 NA NA NA NA
padj
<numeric>
ENST00000539570 NA
ENST00000576455 NA
ENST00000510508 NA
ENST00000474471 NA
ENST00000381700 NA
ENST00000445946 NA